智譜AI在北京成立智算科技公司,強(qiáng)化數(shù)據(jù)處理與存儲(chǔ)支持服務(wù)
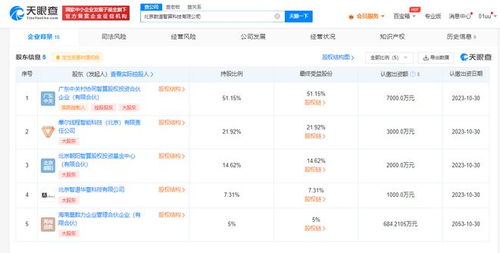
人工智能領(lǐng)域的領(lǐng)軍企業(yè)智譜AI宣布在北京正式成立智算科技有限公司,專注于提供高效的數(shù)據(jù)處理與存儲(chǔ)支持服務(wù)。這一舉措標(biāo)志著智譜AI在算力基礎(chǔ)設(shè)施布局上的重要拓展,旨在為人工智能技術(shù)的研發(fā)和應(yīng)用提供強(qiáng)有力的底層支撐。
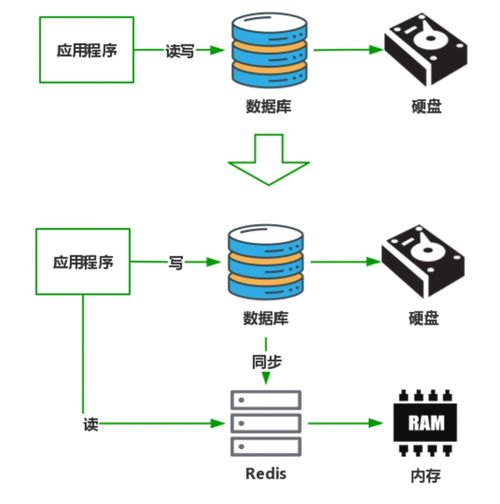
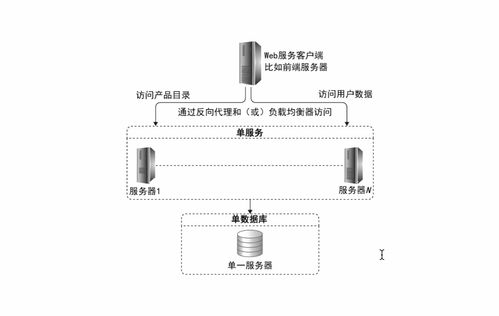
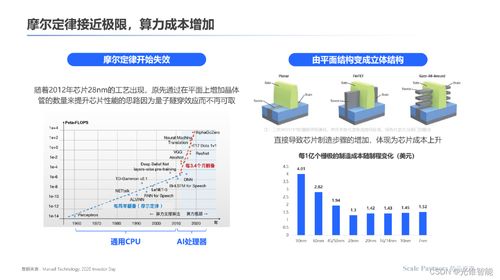
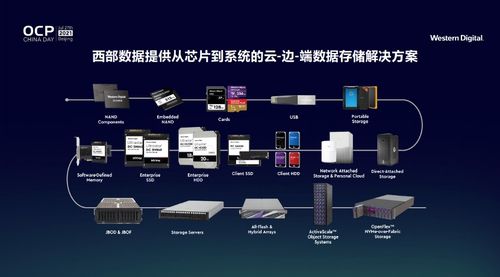
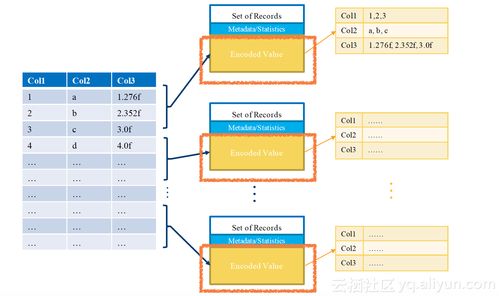
數(shù)據(jù)處理和存儲(chǔ)是人工智能產(chǎn)業(yè)鏈中的關(guān)鍵環(huán)節(jié)。隨著AI模型規(guī)模的不斷擴(kuò)大和應(yīng)用場景的深入,對(duì)大規(guī)模、高性能數(shù)據(jù)處理和存儲(chǔ)能力的需求日益增長。智算科技公司的成立,將依托智譜AI在AI大模型領(lǐng)域的深厚技術(shù)積累,為企業(yè)客戶提供從數(shù)據(jù)采集、清洗到存儲(chǔ)管理的一站式解決方案,助力客戶優(yōu)化計(jì)算資源利用效率,降低運(yùn)營成本。
智算科技公司還將聚焦于智能算力服務(wù)的創(chuàng)新,結(jié)合云計(jì)算和邊緣計(jì)算技術(shù),面向科研機(jī)構(gòu)、互聯(lián)網(wǎng)企業(yè)及傳統(tǒng)行業(yè)用戶,提供定制化的數(shù)據(jù)處理與存儲(chǔ)服務(wù)。通過構(gòu)建高性能的數(shù)據(jù)基礎(chǔ)設(shè)施,智算科技有望推動(dòng)AI技術(shù)在醫(yī)療、金融、教育等領(lǐng)域的落地應(yīng)用,加速產(chǎn)業(yè)智能化轉(zhuǎn)型。
智譜AI此次在北京設(shè)立智算科技公司,不僅體現(xiàn)了其對(duì)算力基礎(chǔ)設(shè)施戰(zhàn)略價(jià)值的重視,也展現(xiàn)了其在AI生態(tài)建設(shè)上的長遠(yuǎn)布局。隨著數(shù)據(jù)處理與存儲(chǔ)技術(shù)的持續(xù)升級(jí),智算科技或?qū)⒊蔀橹沃袊斯ぶ悄墚a(chǎn)業(yè)發(fā)展的重要力量。
如若轉(zhuǎn)載,請(qǐng)注明出處:http://www.amphoraaromatics.cn/product/22.html
更新時(shí)間:2026-06-19 15:53:34